
- >
- Editors
- >
- >
- Attribute Editor
Editor: Attribute Editor ()

Editor: Attribute Editor ()
This tool allows you to edit attributes of groups contained in a Molecule data object. It has additional tabs containing interfaces for exporting attributes to a file or importing them from a file into the data object. We will start with a general description of the attribute concept of followed by a detailed description of the three different tabs.
The Level and Attribute Concept of
In , each molecule consists of several grouping levels, which are chemical subdivisions of the molecule of different degrees of complexity. The levels are ordered in a hierarchy in the way they depend on each other. The most basic subdivision, and therefore the root of the hierarchical tree, is the atom level. All bonds or residues are subdivisions which contain sets of atoms, while secondary structures consist of sets of groups of the residues level, and so on. In the language of database systems, levels are entities and groups are instances of these entities. Figure 1 shows the entity relationship model for the most common grouping levels.

Figure 1: Entity relationship model of the most common levels Each level has a set of attributes. Attributes are properties of groups of type string, integer, or float. The number of levels and attributes depends on the file format from which the molecule is read.
The Edit Tab
The edit tab of the attribute editor (Figure 2) offers two different tools:
- addition, deletion, or renaming of attributes from a level
- changing of attribute values of certain groups of a level
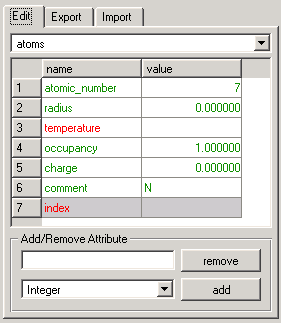
Figure 2: Attribute Editor The pull-down widget at the top of the window lets you choose the level to which the action will be applied. The table below will show all attribute names of the level in the left column. The second column will contain the corresponding attribute values of all completely selected groups of the given level.
The coloration of these table cells can change depending on the attribute and the selected groups:
- gray background: attribute cannot be changed or deleted (attribute is used as an index by )
- white background: attribute can be changed and deleted
- black text: only one group of the level is selected
- green text: several groups of the level are selected but all of their attribute values are equal
- red text: several groups of the level are selected and at least two attribute values of these groups differ (in this case no attribute value will be displayed in the right column)
Changing attributes or attribute values: To change the name of an attribute, simply left-click in the respective cell of the left column and enter the desired name. To change attribute values of the currently selected groups, click in the cell in the right column. Except for index attributes (gray background), all values can be changed. If several groups are selected and no value is displayed (because some values differ), the adjustment of the value in the empty table cell will reset the value of all groups to the given choice. Therefore, the color of this attribute will change from red to green.
To select groups you can use the selection browser.
Adding and deleting attributes: The lower part of the window lets you add or delete attributes by typing the name into the text box and using the appropriate button. When adding attributes, you also must choose the internal format type (string, integer, or float) with the pull-down menu.
The Export Tab
If you have done calculations in which created attributes as a result, you might want to save these attributes to a file which can be used by other software (for example, statistics packages). This tab gives you a powerful ability to write and format your output.Using a predefined format specification: To export attributes to a file, you must specify the format in the format widget. You can load a predefined format specification by clicking on the Predefined Specification button. We have included some simple specifications for common tasks. You can add your own specification by editing the file share/molecules/exportPredefinitions.cfg in your local directory.
Creating a new format specification: A complete explanation of the concept of the formatting string can be found in the following section. We will start with a simple example which shall give you a first understanding of the idea behind the concept:
%(atoms)%(atoms,charge)will write a file which will contain the charge of each atom. Note that after the last parenthesis you have to enter a whitespace, otherwise all charges will be written without delimiters between them.
Iteration-context: The first thing to think about when writing attributes to a file is to decide which level should be the base level of information. This will be the level over whose groups will be iterated, the 'iteration-context'. Usually this is just the same level as that of the attributes you want to write. However, imagine you want to write the residue names of the residues each atom belongs to. In this case, the iteration context is the level 'atoms' while the attribute is of the level 'residues'. To set the iteration-level, just type %(levelname). In the example given above the iteration-context was the level 'atom'.
Text and attribute output: The text after this iteration-context definition specifies the output for each member of the iteration-level. It can contain two things: specification of attributes and additional text that might contain delimiter characters or keywords needed by the importing function of another program. The attribute specification has the form %(levelname,attributename). The additional text can contain everything (including carriage returns) except the character '%'. In the example above the attribute specification was %(atoms,charge) and the additional text was the trailing whitespace.
Iteration context and level dependency: Another example:
%(residues)%(residues,index) %(chains,index)Results in a table which contains the chain index and the residue index for each residue (after the last parenthesis a carriage return must be entered, otherwise there wouldn't be linefeeds between the individual entries).
The slightly modified example
%(residues)%(residues,index) %(atoms,index)might look okay at first glance, but what is the atom index for each residue? In fact it is undefined as each residue can contain several atoms. This is a direct result of the general concept of level dependency. If groups of a level lev1 can contain groups of level lev2, lev1 is dependent on lev2. Inside of an iteration-context only such levels which depend on the iteration-level or the iteration-level itself may be used.
Ending an iteration context: The iteration-context can be ended by the definition of a new iteration-context or the empty definition %(). The latter enables you to write text between different sections. Example:
ATOM SECTION: %(atoms)ATOM ix=%(atoms,index) z=%(atoms,atomic_number) isInRes=%(residues,index) %()RESIDUE SECTION: %(residues)RESIDUE ix=%(residues,index) name=%(residues,name)of course again ended by a carriage return after the last parenthesis. The %() was needed so that the text 'RESIDUE SECTION' wouldn't be repeated for each member of the 'atoms' iteration-context.
Special attributes: As atom coordinates are not stored as attributes but in an internal data array of the object, they are not directly available. However, you can write them by using %(atoms,coordinates) which will be internally translated from an attribute access to an access on the data array. It will write the x, y, and z coordinates delimited by whitespaces.
Options: The option write selected only will limit each iteration context to those groups which are currently selected. For unselected groups no output will be produced.
The Import Tab
If you want to use results of other programs in , you can import them as attributes of groups. An example is the common task of generating partial charges with a molecular force field tool and then reading them in to examine the results.If you haven't done so yet you should first read the previous section about exporting a file, as the syntax of the export format string can be considered to be a simple form of the syntax of the import format string.
Using a predefined format specification: Just as for exporting attributes you can also load predefined format specification with the Predefined Specifications button. You can edit the specification in the file share/molecules/importPredefinitions.cfg in your local directory.
Range of application: When you want to import an attribute your first task will be to take a look at the file to analyze its structure and to find the information that you need. If the information in the file is in nested structures, it is recommended to transform the file into a simpler format by writing, for example, a Perl script or to write your own internal reader method using .
Most of the files you will encounter however contain information simply delimited by certain characters or keywords or located in certain columns of the file. If the format is quite complicated it is sometimes helpful to preprocess the file via the Unix commands 'egrep' and 'cut' to cut out the information needed.
Iteration-context and attribute specification: To get the idea behind the import format syntax, here is an example for a very simple file format:
%(atoms)%(atoms,charge,float);This code expects that there are as many charge values in the file as the number of atoms in the molecule, each separated by a semicolon.
Thus the format string has the same concept of an iteration-context as the export method. The only new part in the example is the type specification 'float' which is needed if the attribute does not already exist inside . If it does, you can omit the type. Type can be 'integer', 'string', or 'float'. Another difference to the export format is that you can specify only attributes of the same level as the iteration-level.
Skipping characters: If you want to specify that there may be an arbitrary number of characters of some type, you can do so by %"char"*. In the example
%(atoms)%(atoms,charge)%" "*;%" "*would be needed if there may be an arbitrary number of whitespaces between the charge values and the semicolon. You can enter more than one character between the quotes. It will read and skip any character contained in the quotes until the token behind the asterisk is found. If the token is not a literal, but an attribute specification, it will skip the characters until the first occurrence of a token that might be of the same type as attribute. The quoted characters can of course also contain a linefeed. A %* will skip any character.
An often occurring problem is that your information is located in a certain section of the file which is initiated by a certain keyword. To jump to this section simply type %*keyword. The following example could be used for the TRIPOS file format which uses the '@<TRIPOS>ATOM' keyword to initiate its atom block.
%*@<TRIPOS>ATOM %(atoms)%(atoms,index) %(atoms,atomic_symbol,string)%*Another common problem is to skip a token. Consider for example that at the start of each line is some alphanumerical token followed by one or more spaces followed by the attribute you want to read. Typing all characters which can be skipped would be tedious, but you can define ranges by using [char1-char2]. Thus the example looks like
%(atoms)%"[a-z][A-Z][0-9].,_-"*%" "%(atoms,charge,float)%*Note that the range is interpreted as a range of the ASCII positions of the given characters.
Using a file-internal iteration: Take a look at the following example:
%(atoms)ATOM:%" "*%(atoms,index)%" "*%(atoms,charge)%*The defined iteration-context won't be needed because for each charge value we know the atom index it is associated with. Thus, the index has the function of an own file-internal iteration-context. This allows you to import attributes of only a part of the groups contained in the molecule. You will need to specify the iteration context 'atoms', however, to make it clear which parts of the format string constitutes the pattern repeatedly searched for in the file. This however only applies if the index attribute has been read first. If there are other attributes before it in the same iteration context, it will not overrule the context.
Reading information in predefined columns Some file formats (like pdb) do not use delimiter characters, but have predefined columns in which information is located. To access these columns you can add width numbers to the attribute specifications and skip specifications. The format is: %width(levelName,attributeName,attributeType) and %width*.
If the charge attribute you want to read is located between the 50th and 59th column of each line, you could specify the format like this:
%(atoms)%49*%10(atoms,charge)%*followed again by a linefeed. The first 49 columns of each line will be skipped. Then the charge attribute will be read from the next 10 columns and the rest of the line will be skipped.