| Name | Last modified | Size | Description | |
|---|---|---|---|---|
| Parent Directory | - | |||
| gb3.jpg | 2008-03-18 17:46 | 6.4K | ||
| nsp1.jpg | 2008-03-18 17:46 | 7.2K | ||
| flowchart.gif | 2008-03-18 17:46 | 14K | ||
| flowchart01.gif | 2008-03-18 17:46 | 34K | ||
| log.gif | 2008-03-18 17:46 | 6.9K | ||
As described in the paper:
Contact:
shenyang@niddk.nih.gov; bax@nih.gov
|
 |
What is CS-ROSETTA?
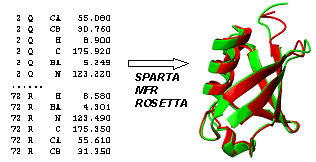
To date, interpretation of isotropic chemical shifts in structural terms is largely based on empirical correlations gained from the mining of protein chemical shifts deposited in the BMRB, in conjunction with the known corresponding 3D structures. Chemical-Shift-ROSETTA (CS-ROSETTA) is a robust protocol to exploit this relation for de novo protein structure generation, using as input parameters the 13Ca, 13Cb, 13C', 15N, 1H and 1HN NMR chemical shifts. These shifts are generally available at the early stage of the traditional NMR structure determination procedure, prior to the collection and analysis of structural restraints. CS-ROSETTA approach, as shown below, utilizes SPARTA-based selection of protein fragments from the PDB, in conjunction with a regular ROSETTA Monte Carlo assembly and relaxation method. Evaluation of 16 proteins, varying in size from 56 to 129 residues yielded full atom models that have 0.7-1.8 angstrom root-mean-square deviations for the backbone atoms relative to the experimentally determined X-ray or NMR structures. The strategy also has been successfully applied in a blind manner to eight structural genomics targets with molecular weights up to 16 kDa, whose conventional NMR structure determination was conducted in parallel. This protocol potentially provides a new direction for high-throughput NMR structure determination, in particular in structural genomics.
Contents
Preparation of input chemical shift table
Identification and exclusion of flexible tails and loops
How to use CS-ROSETTA
Fragment selection
Protein structure generation
Evaluation of CS-ROSETTA models
How to select CS-ROSETTA models
Criteria for convergence and accepting models
Number of models required
Installation files (for Linux/Mac)
| Install.com | CS-ROSETTA Install C-shell Script (last updated @ March 18th, 2008) |
| CSRosetta.tar.Z | CS-ROSETTA main package, scripts, examples (last updated @ March 18th, 2008) |
| PDBH.tar.Z | Database of PDB files, required by MFR/CS-ROSETTA |
| CS.tar.Z | Database of chemical shifts files, required by MFR/CS-ROSETTA |
| ANGLESS.tar.Z | Database of secondary structure classifications and ROSETTA-idealized backbone torsion angles, required by CS-ROSETTA |
Installation
The current implementation of CS-ROSETTA requires the MFR program to perform fragment selection and the ROSETTA program to conduct the protein structure prediction. Therefore, before installing and using CS-ROSETTA package, the newest NMRPipe (incl. the MFR module stored in dyn.tar.Z, mfr.tar.Z, pdbH.tar.Z) (http://spin.niddk.nih.gov/NMRPipe) and ROSETTA programs (http://www.rosettacommons.org/software/) MUST to be installed, as well as other programs such as RCI (http://redpoll.pharmacy.ualberta.ca/download/rci/)and TALOS (http://spin.niddk.nih.gov/NMRPipe/talos/), which are used during data analysis.
Note: If your NMRPipe was obtained and installed before 07/2007, you have to email Frank Delaglio to request the newest NMRPipe in order to run CS-ROSETTAInstallation of ROSETTA
To install the ROSETTA program, users need to (1) register for an academic license at http://www.rosettacommons.org/software/, (2) download (at least) the required packages (current name of the bundle: RosettaBundle-2.2.0.tgz) to the installation directory (for example $ROSETTA_DIR), (3) uncompress RosettaBundle-2.2.0.tgz and all four generated sub-packages (with names of rosetta*.tgz), (4) go to the rosetta++ directory and type "make gcc" (for Linux installation) to compile the ROSETTA source codes. After successful compilation, an executable ROSETTA file with default name "rosetta.gcc" will be generated.
Installation of CS-ROSETTA
Download all above files to the installation directory ($baseDir), type 'install.com' to start the installation. The correctly installed CS-ROSETTA program contains the following contents in the installation directory $baseDir:
|
./com |
CS-ROSETTA scripts directory |
|
bmrb2fasta.com |
Generate a FASTA sequence file from a BMRB chemical shift file |
|
bmrb2talos.com |
Convert a BMRB chemical shift file to TALOS-format |
|
csrosettaInit.com |
CS-ROSETTA initialization script |
|
extract_pdb.com |
Generate PDB full-atom coordinates files from a ROSETTA silent output file |
|
extract_lowscore_decoys.py |
Python script to extract N lowest energy models from a ROSETTA silent output file to a new silent output file |
|
fasta2pdb.com |
Generate a 'dummy'-PDB file from a FASTA sequence file, which is required by MFR as the reference structure |
|
mfr.tcl |
Modified MFR starting script from the original 'mfr.tcl' in NMRPipe, required by CS-ROSETTA |
|
mfr.com |
Conduct MFR fragments search |
|
paths.txt |
Template of "paths.txt" file for ROSETTA |
|
pdb2fasta.com |
Generate a FASTA sequence file from a PDB file |
|
renumber_pdb.com |
Renumber sequence number for a PDB coordinate file |
|
runCSRjob.com |
Starting script to run fragment search and prepare ROSETTA inputs |
|
runRosetta.com |
Template of ROSETTA starting script |
|
runCSRescore.com |
Starting script to re-score ROSETTA full-atom models using experimental chemical shifts |
|
runRosettaRescore.com |
Starting script to extract segments from ROSETTA full-atom models |
|
sparta |
Starting script for program SPARTA |
|
./PDBH |
Directory of PDB coordinates database |
|
./CS |
Directory of SPARTA-assigned chemical shifts database |
|
./ANGLESS |
Directory of secondary structure classification and ROSETTA-idealized backbone torsion angles database |
|
./src |
Directory of supporting C++ program |
|
SPARTA |
Chemical shifts prediction program SPARTA |
|
mfr2rosetta |
C++ program to convert MFR-format fragments to ROSETTA-format |
|
pdbrms |
C++ program to calculate RMSD value between two sets of PDB coordinates |
|
./example |
Directory of a complete example for protein GB3 |
|
./input/gb3.tab |
GB3 input experimental chemical shift table |
|
./input/runCSRgb3.com |
Script for starting GB3 CS-ROSETTA run |
|
./output/ |
All output files obtained by running ./input/runCSRgb3.com |
|
runCSRtest.com |
Script to test CS-ROSETTA installation |
The initialization script csrosettaInit.com includes the definitions for all environmental variables required by CS-ROSETTA. NOTE that the variables $rosettaDir and $csrosettaDir defined in csrosettaInit.com:
setenv rosettaDir /home/software/ROSETTA setenv csrosettaDir /home/software/CSROSETTA
MUST be replaced by users according to their ROSETTA and CS-ROSETTA installation directories.
In order to use the package, users MUST first execute above initialization script, for instance by adding the following command to their .cshrc file:
if (-e $baseDir/com/csrosettaInit.com) then
source $baseDir/com/csrosettaInit.com
endif
If the package is installed successfully and environmental variables are set up correctly, script in the GB3 example directory (examples/runCSRTest.com) will work successfully.
Preparation of input chemical shift table
CS-ROSETTA utilizes the protein backbone 15N, 1HN, 1HA, 13CA, 13CB and 13C'
chemical shifts as inputs and search a structural database for best matched
fragments. The chemical shifts need to be in TALOS format, as defined at
http://spin.niddk.nih.gov/NMRPipe/talos/#preparing shifts. If starting from
a standard BMRB format chemical shift file, a C-shell script
$baseDir/com/brmb2talos.com
can be used to generate a TALOS-format chemical shift file using:
brmb2talos.com bmrbCS.str > inCS.tabAn example of the chemical shifts input format can also be found in the file $baseDir/examples/input/gb3.tab:
DATA SEQUENCE MQYKLVINGK TLKGETTTKA VDAETAEKAF KQYANDNGVD GVWTYDDATK DATA SEQUENCE TFTVTE VARS RESID RESNAME ATOMNAME SHIFT FORMAT %4d %1s %4s %8.3f 1 M CA 54.519 1 M CB 29.320 1 M HA 4.189 2 Q C 174.318 2 Q CA 55.632 2 Q CB 30.865 2 Q HN 8.347 2 Q HA 5.109 2 Q N 123.775 ...Note that the missing chemical shift data are allowed, but the amino acid sequence shown in the header MUST be the full sequence of the protein and MUST start from residue 1, CS-ROSETTA will generate protein structures with the sequence defined in the header.
Residues from the disordered tails and loops are identified from the input
chemical shifts and the criteria in the paper:
(1) S2 < 0.7 from the RCI analysis, and/or
(2) no "good" predictions using the program TALOS
Positively identified flexible residues in the N- and C-terminal tails should be
excluded from the structure prediction by preparing a 'truncated' input chemical
shift table file excluding data (both sequence and chemical shifts) from those
residues. For long flexible loops, it is advantageous to exclude all terms from
the ROSETTA full-atom energy and the chemical shifts rescoring (see details in
Evaluation of CS-ROSETTA models)
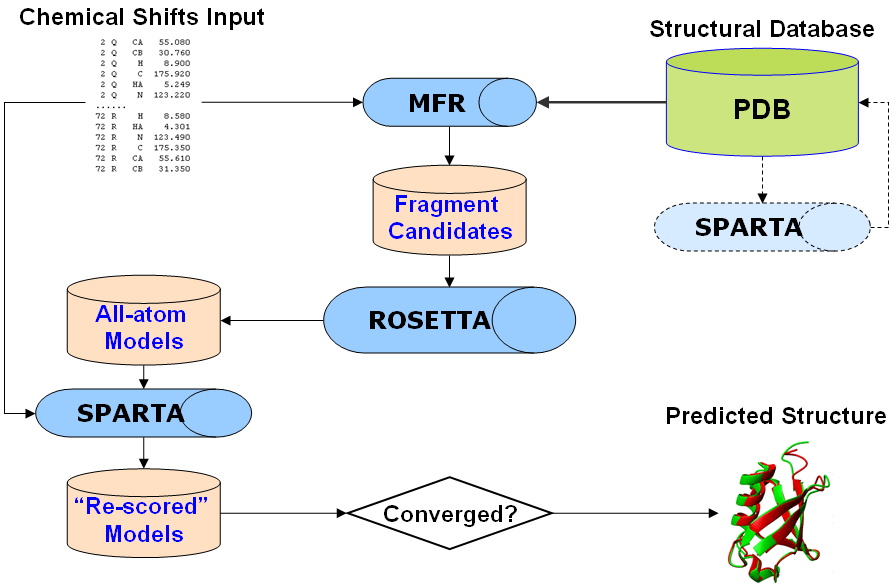
CS-ROSETTA has been designed to work in a black-box manner, and is supported by
multiple C-shell/C++/python scripts/programs. To use CS-ROSETTA for protein
structure prediction, users only require to: (1) run master script
runCSRjob.com
(provided with an input experimental chemical shift file) to select fragments
from the structure database and to prepare a starting package for the subsequent
ROSETTA structure generation, (2) run runRosetta.com (prepared in the previous
step) to perform ROSETTA structure prediction and generate full-atom models, (3)
run runCSrescore.com to add a chemical shift term to the CS-ROSETTA full-atom
"energies". A flow diagram of the CS-ROSETTA protocol implemented in this package looks as shown below:
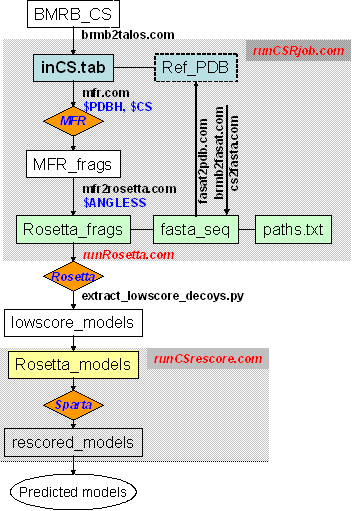
1. Fragment selection - runCSRjob.com
The script runCSRjob.com is the master script to generate MFR fragments and
inputs for ROSETTA. To use this script, an input chemical shift filename MUST be
specified in a command line such as:
runCSRjob.com inCS.tabThe following optional variables are hard-coded in script runCSRjob.com:
set OUTPUT_DIR = rosetta set MFR_EXCL = "PDB1_ PDB2A PDB3X" set PDB_NAME = t000 set CHAIN = _ set TAG = aa set N_MODEL = 5000where $OUTPUT_DIR is the output directory of the fragments and scripts for running ROSETTA (or the ROSETTA running directory), $MFR_EXCL is the list of the proteins (4-letters's PDB name plus 1-letter chain ID) with homologous sequence and known structural homologs to the target protein, for which users like to exclude from the MFR structural database prior to MFR fragment searching, $PDB_NAME and $CHAIN are the 4-letters and 1-letter dummy names indicating the protein name and chain ID, respectively, $TAG is a 2-letters dummy tag (used by ROSETTA), $N_MODEL is the total number of models to be predicted.
The output of running this script will be the MFR-selected 9-residue and 3-residue fragments in ROSETTA format (with file names of $OUTPUT_DIR/$TAG$PDB_NAME$CHAIN09_05.200_v1_3 and $OUTPUT_DIR/$TAG$PDB_NAME$CHAIN03_05.200_v1_3, respectively), a FASTA sequence file ($OUTPUT_DIR/$PDB_NAME$CHAIN.fasta), a ROSETTA paths definition file ($OUTPUT_DIR/paths.txt) and a script file ($OUTPUT_DIR/runRosetta.com) for starting ROSETTA structure generation.
It is recommended for users to read through and understand this script, which would facilitate use of the MFR program and other supporting scripts, and solve any problems during the MFR search (for example, by checking the intermediate and log files). This script (1) Prepares input for the MFR program, (2) Runs the MFR fragment search, (3) Convert fragments to ROSETTA format and (4) Prepares inputs and scripts for running ROSETTA structure generation. In details:
(1) Prepare MFR inputs
The current MFR program in NMRPipe requires a TALOS format chemical shift table
file as input (see Section of "Preparing of chemical shift table"). A "reference PDB" coordinate file is needed to provide the MFR program with the protein's
amino acid sequence. For this purpose, runCSRjob.com creates a dummy "reference
PDB" coordinate file from its FASTA sequence using:
fasta2pdb protein.fasta > dummy_ref.pdb
(2) Run MFR fragment search
The script runCSRjob.com then conducts a MFR fragment search following a command
line of:
mfr.com -cs inCS.tab -ref ref.pdb -out out.tab -excl $MFR_EXCL >& log
where $MFR_EXCL is a list of protein names (without .pdb suffix) that users like to exclude from the MFR structure database prior to the fragment search. MFR fragment selection from searching the structural database is a time-consuming job (1-3 hours); the 'log' file can be used to check the progress.
(3) Convert fragments to ROSETTA format
The MFR-selected 9-residues and 3-residues fragment candidates, generated after
running the MFR fragment search, have file names of the types
frag9.$PDB_NAME.mfr.tab and
frag3.$PDB_NAME.mfr.tab, respectively. Script
mfr2rosetta.com can be utilized to convert these fragments to standard ROSETTA
format:
mfr2rosetta.com -mfr frag9.$PDB_NAME.mfr.tab -segLength 9 > frag9.$PDB_NAME.rosetta.tab mfr2rosetta.com -mfr frag3.$PDB_NAME.mfr.tab -segLength 3 > frag3.$PDB_NAME.rosetta.tab
(4) Prepare ROSETTA running package
runCSRjob.com generates at the last step a new directory (defined by variable $OUTPUT_DIR) and prepares the following required inputs and script for running ROSETTA structure generation:
9-residues and 3-residues fragment files $TAG$PDB_NAME$CHAIN09_05.200_v1_3 and $TAG$PDB_NAME$CHAIN03_05.200_v1_3
FASTA sequence file $PDB_NAME$CHAIN.fasta
ROSETTA paths definition file paths.txt
starting script file runRosetta.com for ROSETTA structure generation
2. Protein structure generation - runRosetta.com
Script runRosetta.com
generated by running the script runCSRjob.com can be
used to start the standard ROSETTA structure generation (fragment assembly and
full atom relaxation). This script includes solely a standard ROSETTA command
line defining the inputs names, the parameters for fragment assembly and
full-atom relaxation
${rosetta} $TAG $PDB_NAME $CHAIN -silent -output_silent_gz -increase_cycles 10 -new_centroid_packing
-abrelax -output_chi_silent -stringent_relax -vary_omega -omega_weight 0.5 -farlx -ex1 -ex2
-termini -short_range_hb_weight 0.50 -long_range_hb_weight 1.0 -no_filters -rg_reweight 0.5
-rsd_wt_helix 0.5 -rsd_wt_loop 0.5 -output_all -accept_all -do_farlx_checkpointing -relax_score_filter
-record_irms_before_relax -acceptance_rate 1.0 -filter1a 10000 -filter1b 10000 -nstruct $N_MODEL
where $TAG,
$PDB_NAME,
$CHAIN (variables for protein identities) and
$N_MODEL
(total number of predicted models) are automatically replaced by their values
defined in script runCSRjob.com.
Running script runRosetta.com in the working directory (where the selected fragments, FASTA sequence and paths.txt are stored) will start the standard ROSETTA structure prediction. The output of the ROSETTA run is a so-called silent output file with name $TAG$PDB_NAME.out, which includes the scores and full-atom descriptions for all accepted models.
The ROSETTA is intensively computationally demanding (in the order of 5-10 minutes per model). Therefore, use of computer clusters is highly recommended. In order to run multiple ROSETTA jobs in parallel for a given project, users can simply run the same script runRosetta.com for each 'parallel' job in the same working directory; ROSETTA will output the results from each job to a single silent output file.
3. Evaluation of CS-ROSETTA models - runCSrescore.com
A ROSETTA silent output file $TAG$PDB_NAME.out contains a header in its first
two lines (first line: the sequence information, second line: definition of
scores and values), and for each model, the scores and values defined in the
header (first line) and the residue-specific description of the full-atom model.
An example ROSETTA silent output file is shown below:
SEQUENCE: MQYKLVINGKTLKGETTTKAVDAETAEKAFKQYANDNGVDGVWTYDDATKTFTVTE SCORE: score env pair vdw hs ss sheet cb rsigma hb_srbb hb_lrbb rg co contact rama bk_tot fa_atr fa_rep fa_sol h2o_sol hbsc fa_dun fa_intra fa_pair fa_plane fa_prob fa_h2o h2o_hb gsolt sasa omega_sc description SCORE: -116.07 -12.24 -1.62 0.31 -3.25 -72.74 0.34 15.99 -28.93 -18.75 -29.62 11.42 17.69 0.00 -13.19 -134.45 -165.57 7.34 84.55 0.00 -3.13 22.82 0.07 -3.11 0.00 -15.74 0.00 0.00 45.19 3958.98 7.55 S_0001_9019 1 E -81.616 95.109 -180.850 0.000 0.000 0.000 -60.979 -52.601 -70.896 0.000 S_0001_9019 2 E -103.775 140.428 181.156 0.662 1.438 3.457 -61.314 178.939 7.568 0.000 S_0001_9019 3 E -128.914 145.607 180.081 -1.325 0.472 6.551 -62.403 85.133 0.000 0.000 S_0001_9019 ...Before rescoring ROSETTA full-atom models using the experimental chemical shift input, it is recommended to extract the models with low ROSETTA energy and 'discard' those 'bad' models with high ROSETTA energy. This expedites further analysis and can be performed by a simple command line:
extract_lowscore_decoys.py silent_file.out N > new_silent_file.outwhere the script extract_lowscore_decoys.py (from whip.bakerlab.org) is used to extract the N lowest-energy models (the energy of a model is defined as the first number in the SCORE line of a silent output file) from a silent file. The output is a new silent output file containing only the selected lowest-energy models.
If residues in flexible loops are positively identified (e.g., by RCI analysis),
the energy terms for those residues should be excluded from the ROSETTA
full-atom energy terms. Therefore, full-atom energies of models in a ROSETTA
silent output file could be rescored by a script
runRosettaRescore.com:
runRosettaRescore.com silent_file.out ref.pdbwhich basically run a single standard ROSETTA command line:
${rosetta} -extract_segment -segments seg.txt -n ref.pdb -s silent_file.out -fullatom -fa_input -all -termini
where "ref.pdb" is a reference
PDB coordinate file (which is required for running standard ROSETTA
"-extract_segment" module for
calculate the C-alpha RMSD values; for instance, the full atom PDB coordinate file
extracted from the lowest energy model in
silent_file.out can be used, see
"Full-atom PDB coordinates extraction"), "seg.txt" is a text file deifining the segment for which ROSETTA
will calculate the new full-atom energies. Here is an example of "seg.txt":6 60 80 99 120 130which tells ROSETTA that only residues 6-60, 80-89 and 120-130 are kept for calculating the new full-atom energy for models, this file MUST locate in the current directory. The output is a new ROSETTA silent file with file name of silent_file_segment.out.
The script
runCSrescore.com is able to be used to apply the chemical shift based
rescoring for ROSETTA full-atom models, using the following command line:
runCSrescore.com silent_file.out inCS.tabwhere inCS.tab is the initial experimental chemical shift input file (the same name as in the variable $CS_NAME defined in script runCSRjob.com). This script:
Details of each step include:
(1) Full-atom PDB coordinates extraction
To rescore the ROSETTA full-atom models, the full-atoms PDB coordinates are
required to be 'extracted' using the full-atom description encoded in the silent
output file, which can be done by using script
extract_pdb.com and the following
command line:
extract_pdb.com silent_file.out
This script actually runs the following ROSETTA command to generate full-atom
PDB coordinates from the information in silent output file 'silent_file.out':
rosetta -extract -s silent_file.out -fa_input -all -termini -write_atoms_only
Note that the generated PDB coordinates will be stored in the directory defined
in paths.txt
OUTPUT PATHS: movie ./ pdb ./
(2) Calculate predicted chemical shifts from ROSETTA full-atom models
The ROSETTA full atom models are evaluated by comparing the initial chemical
shift inputs with chemical shifts predicted for the models by SPARTA. SPARTA
takes the standard PDB coordinates for proteins and predicts the backbone 15N,
1HN, 1H , 13C , 13C and 13C' chemical shifts using the following command line:
sparta -in inPDB.pdb -ref inCS.tab
where inPDB.pdb contains the input PDB coordinates file, inCS.tab is the input experimental chemical shifts (the same input as used for the MFR fragment search). The output is a table file with predicted chemical shifts, and the chi2 value between the predicted and experimental chemical shifts is calculated.
The script runCS_rescore.com runs SPARTA chemical shift prediction for each set of full-atom PDB coordinates generated above, and stores all the outputs in a default directory "./pred".
(3) Collect energy score and apply correction using chemical shifts chi2 value
For each full-atom model, its name and raw ROSETTA full-atom energy score are
extracted from the ROSETTA silent output file
silent_file.out and stored in a
file "name.rawscore.txt". Next, the energy scores will be 'corrected' by using
the corresponding chemical shifts chi2 values stored in a file "CS_chi2.txt".
The new file with the model names and re-scored energy will be "name.rescore.txt",
which for example contains the following contents:
# name raw_energy chi2 rescore_energy S_0001_9019 -116.07 251.854 -53.1065 S_0002_6198 -124.99 140.1 -89.965 S_0003_5600 -123.80 115.019 -95.0452 S_0004_2308 -118.78 148.107 -81.7533 S_0005_3837 -111.47 75.3417 -92.6346 S_0006_9327 -119.05 110.179 -91.5052 ...
(4) Calculate RMSD values to the lowest energy model
The C_alpha RMSD values between each model and the model with the lowest re-scored
energy are calculated using the script pdbrms.
pdbrms is a program written in
C++ and can be used to calculate the RMSD values between one PDB coordinate
and a set of protein PDB coordinates. For example, to calculate the C-alpha RMSD between
ref.pdb and 1.pdb, 2.pdb, 3.pdb,
the following command line can be used:
pdbrms ref.pdb 1.pdb 2.pdb 3.pdb
The script runCS_rescore.com will identify the model with the lowest rescored
energy, and calculate the C_alpha RMSD values to this model for all models in the
./output directory. The final output file "name.rescore.txt" will contain the
model names, C-alpha RMSD values and re-scored energies:
# name rmsd rescored_energy S_0001_9019 1.883 -53.1065 S_0002_6198 1.384 -89.965 S_0003_5600 0.984 -95.0452 S_0004_2308 2.788 -81.7533 S_0005_3837 2.718 -92.6346 S_0006_9327 1.943 -91.5052 ...
By default, the ROSETTA running directory (./rosetta) contains the following contents upon finishing the above CS-ROSETTA protein structure generation:
| aat000_09_05.200_v1_3 | 9-residues fragment input for ROSETTA |
| aat000_09_05.200_v1_3 | 3-residues fragment input for ROSETTA |
| t000_.fasta | FASTA sequence |
| paths.txt | ROSETTA paths definition file |
| aat000.out | ROSETTA output silent file |
| ./output | Directory for full-atom PDB coordinates |
|
S_*.pdb |
Extracted full-atom PDB coordinates |
|
pred |
Directory for SPARTA chemical shifts prediction summaries |
| name_rawscore.txt | Table file for model names and raw ROSETTA full-atom energies |
| CS_chi2.txt | Table file for chi2 values of the SPARTA-predicted and experimental chemical shifts |
| name_rescore.txt | Table file for model names and re-scored ROSETTA full-atom energies |
| rms2LowRawscore.txt | Table file for model names and RMSD values relative the model with the lowest raw energy |
| rms2LowRescore.txt | Table file for model names and RMSD values relative the model with the lowest re-scored energy |
How to select CS-ROSETTA models
Criteria for convergence and accepting models
After finishing CS-ROSETTA structure generation, users have to decide whether the ROSETTA models are acceptable. For this purpose, it is convenient to plot the "landscape" of (re-scored) ROSETTA full-atom energies of all models with respect to their C_alpha RMSD values relative to the lowest-energy model, using the data stored in a file "name.rescore.txt".
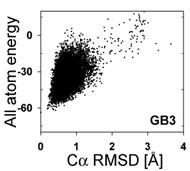
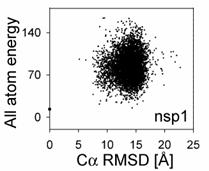
By using the current method implemented in CS-ROSETTA package, 5,000 to 20,000 predicted CS-ROSETTA models are generally required to obtain the convergence. For small proteins (<= 90-100 amino acids), 1,000 to 5,000 predicted CS-ROSETTA models often sufficient. ROSETTA takes about 5-10 minutes to calculate one all-atom model on a single 2.4GHz CPU.