
- >
- Editors
- >
- >
- Molecule Editor
Editor: Molecule Editor ()

Editor: Molecule Editor ()
This tool can be used to change the geometry and topology of a Molecule data object. To change group attributes, you can use the Attribute Editor.Most of the editor's functions are applied to the set if selected atoms, Atoms can be selected by using the selection browser or by directly clicking on atoms in the viewer.
The molecule editor, Figure 1, has three tabs:
- The Transform tab contains all tools to change the geometry of selected atoms by adjusting positions, bond lengths, torsional angles, and bond angles.
- The Tools tab offers methods to split or copy parts of the molecule, as well as an interface for adding or removing bonds between atoms.
- The Building tab offers methods to add or remove atoms, bonds and groups, or to change their chemical properties.
Additionally several menus exists to manipulate the display, assign or change chemical properties and to create new levels or attributes.
Preparing the Molecule
Many of the editing methods of the Molecule Editor need to compute chemical properties of the moleculem which are derived from fixed properties representing the atomic configuration, namely the valency of the atoms, its atomic number and its formal charge. The atomic number is a mandatory attribute of the atom level and the valency is represented as explicit bonds in the molecular data structure and as an additional number of implicit hydrogens.The implicit hydrogen number and the formal charge can be supplied as attributes of the atom level. When the editor starts, this information will be read from the formal_charge and implicit_hnum attributes. If no such attribute exists, they are set to 0 for each atom. Often this is incorrect. PDB files for example usually don't contain hydrogens and the number of implicit hydrogens therefore needs to be adjusted. It is not possible to generate such missing information without further guidance from the user, as the way in which this information needs to be generated depends on how the molecule was generated. The Editor, however, offers a set of tools which allow generating this information easily for most typical cases. The tools menu allows the user to assign a standard implicit hydrogen number a implicit hydrogen number and formal charges. For molecules which contain neither a formal_charge attribute nor explicit hydrogens or an implicit_hnum attribute, you need to use the first method. It will assign sufficient hydrogens to each atom to reach its most common valency. After this you may need to adjust valencies and formal charges. If the formal_charge attribute is known, you should use the second method instead. It will consider changes in the standard valencies caused by the electron transfer. If all hydrogens are known, use the third method which will adjust formal charges to reach standard valencies. You can always check these internal properties by enabling them as labels in the 'view' menu.
As mentioned earlier, this information may change during the editing process. When the editor is closed by pressing the OK button the current state will be written into the atom level attributes.
The View Menu
The view menu allows the user to adjust some visualization properties of the MolView that is used by the editor. 'Hide H' will hide or show the hydrogen atoms. Additionally labels can activated which show important atomic properties:
- None: Labels deactivated.
- Index: The internal atom index (counted from 0).
- Standard: Modes formal charge and radicals combined.
- Formal charge
- Implicit H-Num: Number of implicit hydrogens.
- Lone Pairs: Number of lone electron pairs.
- Atom Type: MMFF94[1] atom type. Atom typing for certain atoms may fail for rare atoms or uncommon or incorrect chemical environments.
- Oxidation number
- Radicals: Shows an asterisk for atoms that contain unpaired electrons.
The Tools Menu
- Assign standard implicit hydrogens: Assign the number of implicit hydrogens while not considering any additional data like the formal charge. Use this if the molecule contains neither explicit hydrogens nor formal charges. After using this assignment you will have to adjust formal charges (if there are any) manually.
- Assign implicit H-Num: Assign number of implicit hydrogens while taking into account formal charges. Use this if the molecule contains formal charges, but no explicit hydrogens.
- Assign formal charges: Assigns formal charges while taking into account the number of implicit hydrogens. Use this, if the molecule contains explicit hydrogens but no formal charges.
- Add H: Makes all implicit hydrogens explicit.
- Add Polar H: Makes all implicit hydrogens of polar atoms explicit.
- Strip H: Makes all hydrogens implicit.
- Strip non-polar H: Makes all non-polar hydrogens implicit.
- Strip H20: Removes all water molecules (including unbound O or H atoms and OH molecules).
- Kekulize: Assign a Kekule structure to the molecule, in effect replacing all aromatic bond systems with alternating single and double bonds.
- Kekulize Non-Rings: Assigns a Kekule structure to all non cyclic parts of the molecule. This is necessary for the editor to work properly as aromaticity is only allowed for rings.
- Dekekulize: This will check all rings system for aromaticity, using the extended Hueckel rule and assign aromaticity where appropriate.
Transform Tab
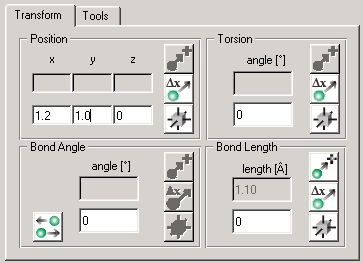
Figure 1: Transform Tab The transform tab is divided into four different sections. Each section can be used to adjust certain coordinates of the currently selected atoms. The different coordinate types are:
- Position is the Cartesian coordinate of an atom. If several atoms are selected, only relative changes are allowed.
- Bond Length is the distance between two selected atoms.
- Bond Angle is the planar angle between three selected atoms.
- Bond Torsion is the dihedral angle between four atoms.
Coordinates can be set absolute or relative to their current value. In each section, the upper row (with gray background) displays the current absolute value. In the lower row (with white background) you can enter a new value. In the right part of each section are three buttons. The first button transforms the coordinate while assuming that the entered value is an absolute value. The second button simply applies the transformation with the entered value text as a relative difference. The third button will activate a dragger in the viewer for adjusting the coordinate interactively. The Molecule Measurement module can be used to display currently edited bonds or angles.
Which buttons can be used and which are disabled depends on the number of currently selected atoms.
An additional toggle button can be found in the Bond Angle section. If this toggle is activated, the interactive adjustment with the dragger will cause both bonds to be bent symmetrically, otherwise only one bond will be bent.
The position dragger can be moved on the plane determined by the face of the cubic dragger you click on. You can additionally rotate the selected groups by dragging the green knobs.
Tools Tab
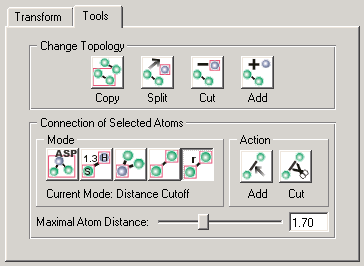
Figure 2: Tools Tab The tools tab is subdivided into two sections:
Change Topology
The cut button will cut the currently selected groups out of the molecule. The split button will do the same but will copy the groups into a new molecule which will be added in the Project View. The copy button will leave the current molecule unchanged while copying the selected groups into a new molecule which will be added to the Project View. When pressing the add button, a window will open which will let you choose another molecule in the Project View whose groups you want to add to the current molecule.
Connection
The Connection section offers different options for influencing the bonding of the currently selected atoms On the right side of the interface are buttons for adding or removing bonds between the currently selected atoms. The set of bonds which will be added or removed will depend on the connection mode that you can choose on the left side of the interface.
- Standard: This is the most reliable method for adding bonds to a protein or DNA/RNA. It will look up all residues in a residue database and add bonds accordingly. This method will only work for molecules that contain the residue type attribute. For connections between different residues it will check all residues on a chain sequentially. When used together with the cut action all bonds will be removed which can not be found in the database.
- Bond length table: This option lets you add bonds between selected atoms by looking up their bond lengths in the file bondLengths.cfg which can be found and edited in your local /share/molecules directory. If the distance of two atoms does not deviate further than a certain threshold from the bond length between the respective elements in the table, the bond will be added. This method is able to distinguish between single, double, triple, and aromatic bonds. It should be the first choice for non-standard residues or molecules not containing residue information. Just like the Standard option this mode can also be used together with the cut action. In this case, all bonds which deviate too strongly from the bond length table will be removed.
- External: This mode can only be used together with the cut action. It will remove all bonds between selected and unselected atoms thus enabling you to disconnect certain parts of the molecule from the rest.
- All: This mode will add or remove all possible bonds between the selected atoms.
- Distance Cutoff: This last mode uses a distance cutoff for deciding which bonds to add or remove. The Maximal Atom Distance slider determines the maximal distance of two bonded atoms in Å. When using this mode together with the add action all bonds between atoms which are nearer than the cutoff distance will be added. Equally, the cut action will remove all bonds whose length is greater than the threshold.
Building
Adding atoms or groups is accomplished by selecting a single atom and replacing it with a new one. Usually, the selected atom would be a hydrogen, but you can replace heavy atoms as well. Hydrogens for the new parts are automatically added.The atoms section contains buttons to add atoms of commonly used elements in organic chemistry. With the '->' button you can activate a periodic table to select less common elements. Other buttons in this section are:
- + and -: Increases or decreases the formal charge of the atom by 1. The number of hydrogens will be automatically adjusted.
- +V and -V: Increases or decreases the valency of the atom by one. They will appear as new or removed explicit or implicit hydrogens. Adding valencies is only possible if the atom has lone electron pairs or an unpaired electron. Removing valencies is only possible, if the atom has explicit or implicit hydrogens.
- Delete: Deletes all selected atoms.
The carbon chains / rings section allows the user to add carbon chains or cyclocarbons. The number of the carbon atoms can be adjusted and the hybridization can be selected, with 'sp2' creating chains of the form C=CC=C... or aromatic rings.
The bonds section contains buttons to add bonds, replace bond order or, delete bonds. You need to select two atoms. Clicking on '-' will create a single, '=' a double, '#' a triple, and '~' an aromatic bond. If the atoms were already connected the bond order will be simply changed. We advise against assigning aromatic bonds by hand. There is no clear definition of what constitutes 'aromaticity' and creating aromatic bonds outside of ring structures can result in some chemo informatics algorithms of the editor returning incorrect result. Instead, always assign Kekule structures. You can then assign aromatic bond orders by using the 'Dekekulize' tool which guarantees that the internal data-structures stay chemically consistent.
Button Group
- OK will close the editor and accept all changes made to the molecule.
- Cancel returns from the editor restoring the last accepted state.
- Reset resets the molecule to the last accepted state without canceling the editor.
- Apply accepts all changes. This means that resetting will return the molecule to the current state.
- Undo undoes the last change. Currently, for efficiency reasons, this is only possible for coordinate transformations of the transform tab.

Figure 3: Button Group of the Molecule Editor
Creating Molecules from Scratch
The editor can only be invoked when a molecule already exists. To create a molecule from scratch you can use the following Tcl command:
newMolFromSmiles <SMILES>which will generate a molecule from a smiles string [2]. If you want to use square brackets '[' and ']' you need to enclose the SMILES in curly brackets to avoid Tcl command substitution. This is also necessary for the bond direction identifier '\' which is usually interpreted as an escape character. Examples:
newMolFromSmiles c1ccnc(C)c1 newMolFromSmiles {C[O-]} newMolFromSmiles {F/C=C\F}
Scripting Interface
Most functions of the MoleculeEditor are scriptable. An editor associated with a molecule may be created with the command
set <myEditor> [ <moleculeObject> createMoleculeEditor ]where <moleculeObject> is the name of the molecule object as it appears in the Project View and myEditor is an arbitrary tcl variable that will contain the name of the created editor object and which can then be used with the following commands:
addH addPolarH removeApolarH removeH removeWater replaceAtomAtomicNum <atomicNumber> replaceAtomSmiles <smilesStr> assignFormalCharge assignHNum assignStandardHNum incCharge incValency decCharge decValency dekekulize kekulize setBondAromatic setBondDouble setBondSingle setBondTriple minimize reset apply copy createAttributeMMFFCharge createAttributeMMFFType createLevelAngles createLevelDihedrals createLevelOOPDihedrals createLevelRingSystems createLevelRings createMMFFParameterization
References
[1] Halgren,T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J.Comp.Chem., 17, 490-519 1996. [2] Weininger,D. 'SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules', J. Chem. Inf. Comput. Sci. 28, 31 - 36 1988.